Nov 10 2025
/
FDA’s Updated Biosimilar Policy: What it really means in the US, EU5, Japan, and China
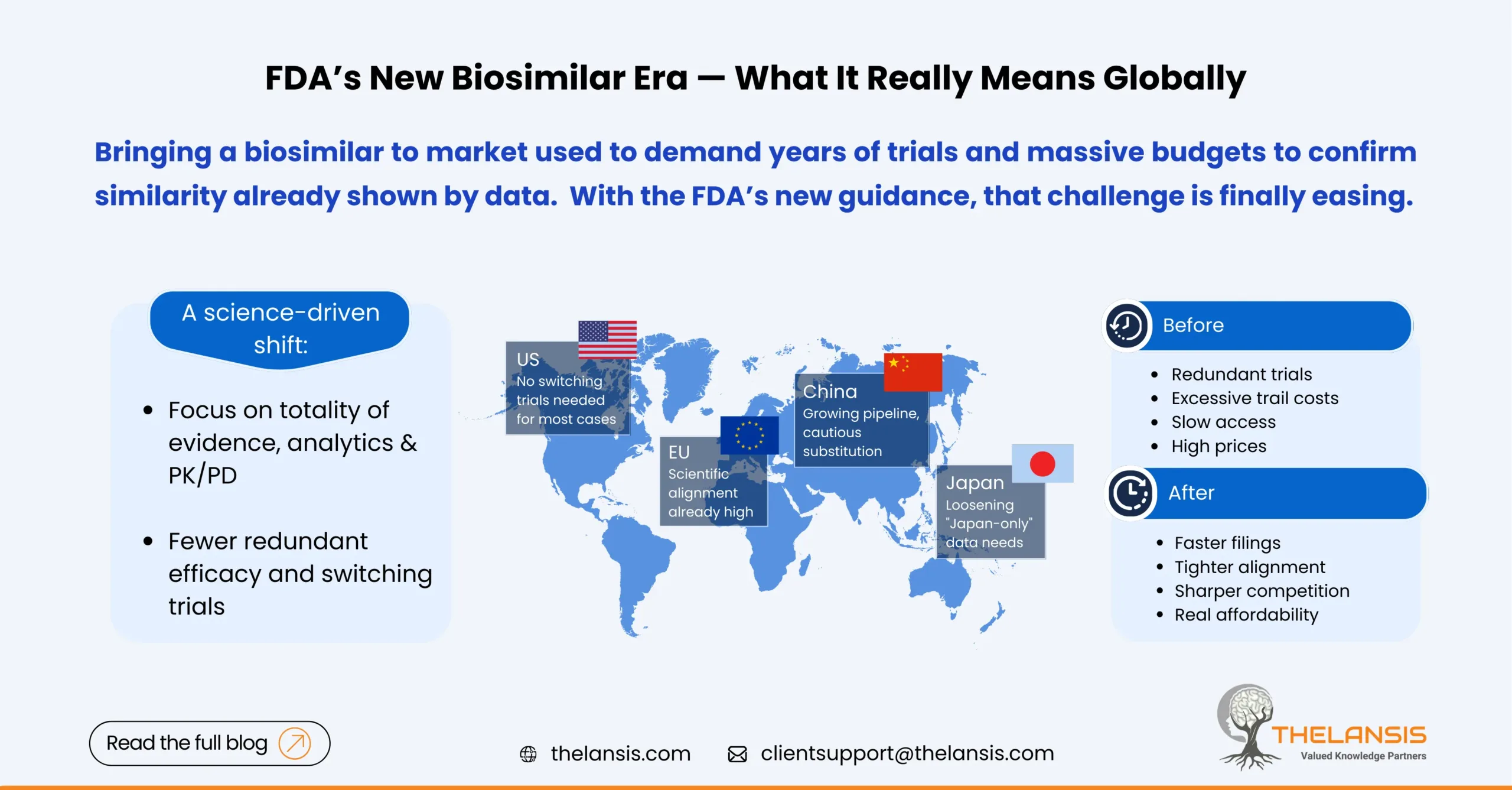
The regulatory landscape for biosimilars is shifting significantly in 2025. The FDA has taken two big steps that collectively make it faster and cheaper to bring many biosimilars to market:
- In June 2024, the FDA proposed to drop “switching” studies for the interchangeable designation, saying most products won’t need extra human trials just to prove patients can switch back and forth.
- In October 2025, the FDA issued new draft guidance signaling that comparative efficacy trials may often be unnecessary for showing biosimilarity, putting more weight on analytics, PK/PD, and totality of evidence. The FDA also previewed steps to make interchangeability more straight forward.
Those two moves won’t solve every commercial hurdle, but they materially reduce development burden.
Below is a breakdown of the key policy changes, what they mean for different players, and how each region might respond.
United States: Simpler files, but access still decides winners
What changed
Interchangeability: The agency updated expectations around switching studies; many products won’t need them. That knocks out a costly step with minimal incremental safety value.
Biosimilarity packages: the new CES draft signals fewer large efficacy trials where state-of-the-art analytics and sensitive PK/PD data already answer the key questions.
Why it matters
Time and cost drop for sponsors; more programs pencil out. FDA’s own data notes numerous “interchangeable” approvals were already granted without extra switching trials, the guidance simply codifies the science.
Interchangeability becomes attainable for more products. Where state laws allow pharmacy substitution, this label can speed uptake as seen with Semglee and Cimerli.
Reality check
Policy ≠ uptake; Even after 9th biosimilar of adalimumab entered, HUMIRA still stands high with more than 80% of market share for much of 2024 and 2025, because of contracting, formulary dynamics, and co-promotion plays. Expect FDA’s policy to help, but payers remain the decisive lever.
The EU5: Already there on interchangeability and it shows
Scientific alignment is already high. Since 2022–2023, the EMA/HMA have stated that EU-approved biosimilars are interchangeable on scientific grounds; countries set their substitution rules. FDA’s 2024/2025 moves largely converge with that stance.
Country-level wrinkles you should factor in:
- Germany: Pharmacy-level switching/substitution rules were operationalized via the G-BA, with new biosimilar switching provisions taking effect March 15, 2024. Plan for region-by-region contracts and “aut idem” mechanics.
- France: Pharmacy substitution exists but is narrow: since 2022, it’s permitted for filgrastim and pegfilgrastim, with broader expansion discussed in the 2024 reforms. Expect substitution to grow, but still check the ANSM lists and local decrees.
- Italy: Automatic substitution is not permitted; AIFA leaves switching to the prescriber. Commercial plans rely on hospital tenders and clinician engagement rather than pharmacy substitution.
- Spain: No automatic substitution in community pharmacies; adoption is driven by regional policies, tenders, and prescriber buy-in.
- United Kingdom: The MHRA/NHS position is clear: once authorised, biosimilars are interchangeable with the reference (and with each other using the same reference). Uptake hinges on NHS procurement and switching programmes.
So what?
For EU5 launches, technical packages can be built once and leveraged on both sides of the Atlantic; the market access plan still needs country-specific tactics (tendering, clinician education, device training).
Japan: less “Japan-only” data, faster alignment
Japan’s PMDA has been revising biosimilar guidance and Q&As, including a 2024 administrative notice and subsequent updates that relax the expectation for Japanese-subject data in every case with appropriate justification, and encourage early consultations on the clinical package without Japanese subjects. That reduces duplicative trials and speeds global alignment.
Takeaway: Anchor on global analytics/PK, plan post-marketing commitments, and engage KOLs to support physician-led switching.
China: Growing pipeline, cautious substitution
China’s NMPA has built out a modern framework (e.g., facilitating one-off import of reference biologics for biosimilar development) and continues to expand approvals, studies count 30+ antibody biosimilars by late 2023. Uptake is increasing but automatic pharmacy substitution is not a national default; switching tends to be a clinical decision, with adoption influenced by volume-based procurement and hospital policies.
Signal to watch: provincial procurement rounds increasingly favor price-competitive biosimilars and accelerating share once listed, mirroring tender dynamics seen in Europe. Policy specifics vary by province; always check the latest VBP notices.
What manufacturers, payers and clinicians should do next?
Manufacturers (all regions)
- Right-size clinical plans: lead with analytics/functional comparability and sensitive PK/PD; add efficacy only if a residual uncertainty truly needs it under FDA’s 2025 CES draft.
- Design for interchangeability from day one (device, instructions, risk management), then map US state substitution laws and EU country substitution rules for launch pull-through.
- Invest in access mechanics: patient cost-share relief, hub services, and specialty-pharmacy pull-through, especially in the US where payer dynamics dominate outcomes.
Payers and procurement bodies
- Translate regulatory savings into patient savings. Zero- or low-copay designs and clear interchange policies move share much faster than policy shifts alone.
- In EU5 and China, tenders with device-training and supply-assurance scoring prevent false economies and support sustained use.
Clinicians and hospital pharmacies
- Lean on EMA/HMA and MHRA statements (EU/UK) and the evolving FDA stance to normalize switching where appropriate; incorporate device training and traceability in SOPs.
Bottom line
The FDA’s 2024/2025 updates lower the trial burden and tighten global alignment with the EU, making it easier to file one core package across the US, EU5 and Japan, while China’s framework keeps maturing. Winners won’t just “save a study”, they’ll reinvest those savings in market access, clinician confidence, and patient affordability—the levers that actually move share.